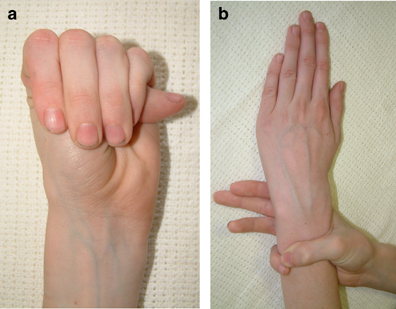
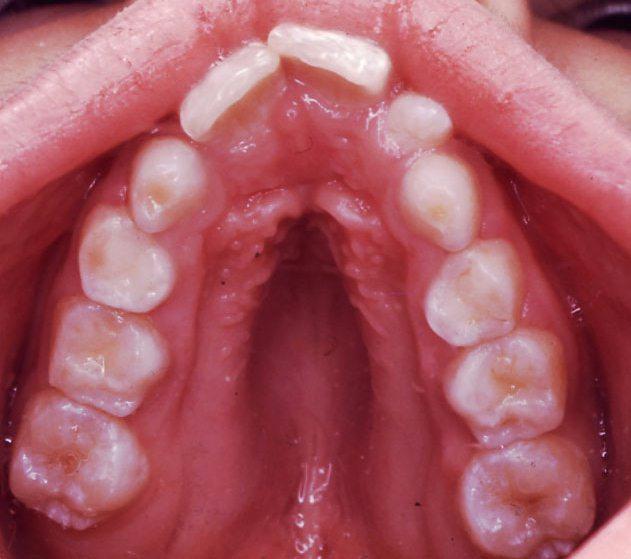
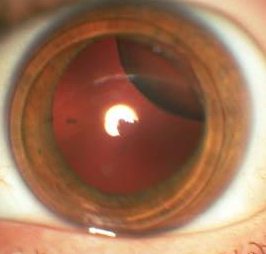
Educating others about genetic disorders allows us all to be more aware of all the different types of people around us, and to not be so insensitive at times. While it's already hard enough to have to deal with the medical issues that Marfan Syndrome brings along, the last thing someone afflicted with the disorder needs is to feel like an outsider from comments and teasing. Simply knowing about disorders such as Marfan Syndrome will stop us from thinking, "What's wrong with that person?" to understanding that they're really just like everyone else... only maybe a bit taller and skinnier.
Simply check out this article about Michelle Smith, now 22 years old, from Scarborough, Texas. Previously a cheerleader and gymnast, she had to give up sports and start having her health monitored after being diagnosed with Marfan Syndrome. But she then turned to new hobbies, such as pageantry and volunteer work, particularly to spreading awareness for the syndrome. The National Marfan Foundation honored her with a Silver Service award, and she even had the opportunity to meet several celebrities.
 |
| Michelle Smith, a beautiful young lady with Marfan Syndrome |
Or ever heard of the popular indie rock band Deerhunter? If you haven't, listen to their song Helicopters. It's great:
The lead singer of the five-piece band is Bradford Cox, and he actually has Marfan Syndrome.
 |
| Deerhunter, with Bradford Cox on the far right |
 |
| Cox preforming at Osheaga 2012 |
While these are just a couple examples of amazing people with Marfan Syndrome, more awareness from the public about Marfan Syndrome will support the many others who have it, and also prevent ignorance. At one point, people even thought that the Olympic swimming champion Michael Phelps had Marfan Syndrome, simply because he was lean and had long arms (here is an article from Fox News that goes further in-depth on the topic.) But anybody who was well-educated on the disorder would know that Marfan Syndrome would prevent Phelps from participating in any swimming activity, because of the dangerous impact it would have on his heart.
Instead of confusing accusations of Marfan Syndrome, people should be able to focus on the actual issue on hand and play a role in aiding medical research so that better treatment can be provided for the individuals that do have the disorder. Check out the Marfan Foundation and the Marfan Trust for possibilities for donations and fundraising. Getting invoved will allow for results that increase early diagnosis, ensure life-saving treatment, and give relentless support to families, caregivers, and healthcare providers.